Amyotrophe Lateralsklerose (ALS) – unheilbare Nervenkrankheit
Die amyotrophe Lateralsklerose (ALS) ist eine fortschreitende Krankheit des zentralen Nervensystems. Sie führt zu Lähmungen, die das Sprechen, Schlucken und Atmen beeinträchtigen. Bisher gibt es keine Heilung, eine entsprechende Therapie kann aber die Symptome lindern und die Lebenserwartung erhöhen.
-

- © Getty Images/herraez
Kurzübersicht: Amyotrophe Lateralsklerose (ALS)
Definition: Die degenerative Erkrankung des zentralen Nervensystems führt zu einer fortschreitenden Zerstörung von Nervenzellen im Gehirn und Rückenmark. Da motorische Nervenzellen (Motoneuronen) betroffen sind, zählt ALS zu den Motoneuronerkrankungen.
Symptome: Nach ersten muskulären Ausfällen kommt es zu massiven Schwierigkeiten beim Schlucken, Sprechen und Atmen sowie im späteren Verlauf zu einer Lähmung der gesamten Körpermuskulatur.
Verlauf: ALS ist unheilbar und führt meist innerhalb von etwa zwei bis fünf Jahren zum Tod.
Therapie: Es gibt Möglichkeiten zur Behandlung, die das Fortschreiten der Krankheit etwas verzögern und die Lebensqualität entscheidend verbessern können.
Artikelinhalte im Überblick:
Was ist ALS?
Die amyotrophe Lateralsklerose (ALS) ist eine fortschreitende Erkrankung, bei der Nervenzellen zugrunde gehen. In der Folge kommt es zu einem zunehmenden Muskelschwund und einer eingeschränkten Bewegungsmöglichkeit.
ALS ist eine Erkrankung der ersten und zweiten Motoneurone. Diese Nervenzellen sind für die Ausführung von willkürlich gesteuerten Bewegungen, also für die Motorik zuständig. Die ersten (oberen) Motoneurone entspringen im Gehirn und führen durch das Rückenmark zu den Wirbelkörpern. Im Rückenmark werden die Signale auf ein zweites (unteres) Motoneuron übertragen, das dann bis zum entsprechenden Muskelansatz weiterverläuft.
-

- © Getty Images/Maria Pilar Martinez Aguerri, Lifeline
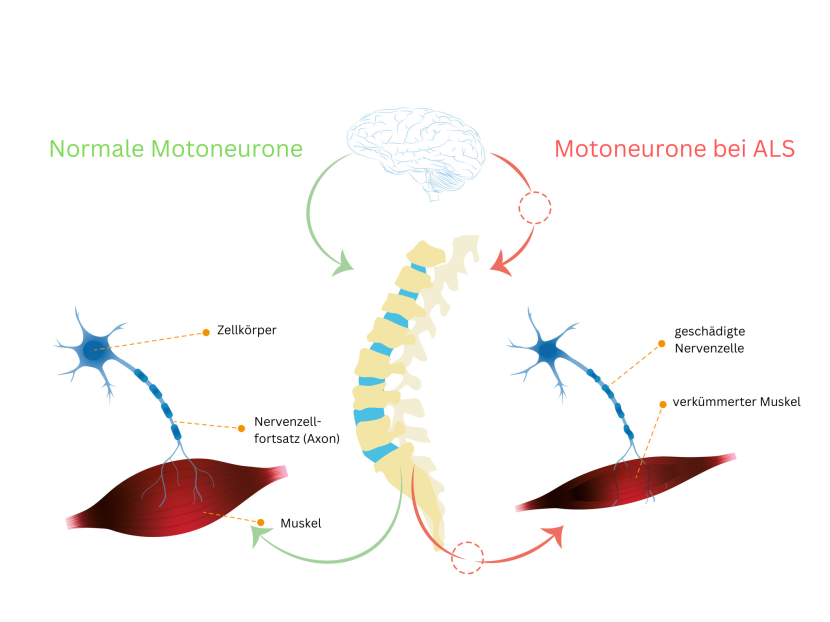
Sonderformen:
primäre Lateralsklerose: Bei dieser Sonderform ist lediglich das erste Motoneuron geschädigt. Sie tritt in jüngeren Jahren auf als die klassische ALS, schreitet sehr viel langsamer voran und verläuft milder. Beim britischen Astrophysiker Stephen Hawking wurde die Nervenkrankheit mit 21 Jahren diagnostiziert, er starb im Alter von 76 Jahren.
progressive spinale Muskelatrophie (PMA): Eine sehr seltene Variante, bei der ausschließlich das zweite Motoneuron betroffen ist. Verlauf und Symptome sind ebenfalls etwas langsamer und milder.
ALS mit frontotemporaler Demenz (ALS-FTD): Hier werden frühzeitig die Neurone im Frontallappen im Gehirn angegriffen. Bei diesen Personen kommt es oft zu Verhaltensauffälligkeiten und Persönlichkeitsveränderungen.
Häufigkeit der ALS
Die amyotrophe Lateralsklerose gilt zwar als häufigste Form der Motoneuronerkrankungen, tritt aber dennoch relativ selten auf. Pro Jahr erkranken in Deutschland zwei von 100.000 Menschen. Der Erkrankungsgipfel liegt um das 60. Lebensjahr herum. Männer sind insgesamt etwas häufiger betroffen als Frauen.
Im Frühstadium sind Symptome der ALS unauffällig
Die Krankheit beginnt schleichend und unauffällig. Als erste Anzeichen fallen den Betroffenen meist Ungeschicklichkeiten auf. Sie stolpern häufiger als sonst oder lassen etwas fallen.
Mögliche Beschwerden:
- asymmetrische Muskelschwäche
- geringe Belastbarkeit der Muskulatur
- allgemeine Müdigkeit
- schmerzhafte Muskelkrämpfe
- Sprechstörungen (Dysarthrie)
- Schluckstörungen (Dysphagie)
Allerdings können die genannten Symptome auf viele verschiedene Erkrankungen hinweisen. Wesentlich wahrscheinlicher sind als Auslöser nervliche Störungen wie beispielsweise ein Bandscheibenvorfall, eine Myopathie oder auch ein Schlaganfall (Apoplex).
Symptome und Komplikationen im fortgeschrittenen Stadium
Die Symptome breiten sich fast immer kontinuierlich aus und führen dann zum typischen Vollbild einer klassischen ALS: schmerzlose Lähmungen am ganzen Körper in Kombination mit spastischen (krampfartigen) Beschwerden in unterschiedlichen Ausprägungen.
Eine Ausnahme bilden der Herzmuskel, die Augenmuskeln und die Schließmuskeln von Blase und Darm, die bei ALS im Normalfall nicht betroffen sind.
Häufig kommt es im fortgeschrittenen Stadium zu unwillkürlichen Muskelzuckungen. Sie sind normalweise nicht schmerzhaft. Fast immer resultieren die Spasmen irgendwann in einer vollständigen Lähmung der Skelettmuskulatur (Tetraplegie) und dem totalen Verlust aller motorischen Fähigkeiten.
Mögliche Symptome im Verlauf einer ALS:
verlangsamte Atmung und Atemaussetzer
Schlafstörungen aufgrund nächtlicher Atemprobleme
Müdigkeit, Konzentrationsprobleme und Kopfschmerzen
Sekretansammlungen in den Atemwegen
Atemwegsinfekte und Lungenentzündung (Pneumonie)
Schluckstörungen, Probleme mit dem Kauen, Essensverweigerung und Gewichtsverlust
Ausbreitung der Lähmung (Parese) mit vollständigem Verlust der Muskelbeweglichkeit
Muskelschwund (Muskelatrophie)
unwillkürliche Zuckungen von Muskeln (Faszikulationen)
pathologisches Lachen, Gähnen oder Weinen, obwohl Betroffenen nicht danach ist
Da es sich um eine Erkrankungen der motorischen Nerven handelt, gehören Empfindungs- und Sensibilitätsstörungen normalerweise nicht zu den Symptomen. Auch die kognitiven Funktionen sind bei der klassischen ALS lange Zeit kaum eingeschränkt. Patient*innen behalten häufig bis zum Ende einen wachen, funktionierenden Verstand.
Ursachen für ALS sind weitgehend unbekannt
Warum es zur Entwicklung einer ALS kommt, ist nach wie vor nicht geklärt. Nur bei wenigen Betroffenen liegt eine erbliche Veranlagung vor.
In über 90 Prozent der Fälle tritt die Krankheit ohne erkennbare Ursache auf. Diese Form wird als sporadische ALS bezeichnet.
Bei etwa 10 Prozent der Betroffenen lässt sich eine familiäre Häufung feststellen, was für eine Genmutation spricht. Dabei kommen mindestens sieben verschiedene Gene als Auslöser in Frage, meistens liegt eine Kombination mehrerer Gendefekte vor.
Auf einigen Inseln im Westpazifik tritt die ALS besonders häufig auf. Sie nimmt dabei auch einen unterschiedlichen Verlauf mit fortschreitender Demenz und Parkinson-Symptomen. Diese endemische Form ist als Western Pacific ALS oder ALS PD complex bekannt.
Diagnose durch Ausschluss anderer Erkrankungen
Bei Verdacht auf ALS stehen Ärzt*innen neurologische Untersuchungsmethoden und bildgebende Verfahren zur Verfügung. Erst wenn diese Untersuchungen eindeutig eine Schädigung des ersten und zweiten Motoneurons ergeben haben, sprechen Fachleute von ALS.
Zunächst müssen Krankheiten mit ähnlichen Symptomen sicher ausgeschlossen werden. Hierfür kommen beispielsweise verschiedene Nervenkrankheiten wie Polyneurosen, Muskelerkrankungen, Autoimmunerkrankungen oder auch ein Bandscheibenvorfall infrage.
Mögliche Untersuchungen:
Elektromyographie (EMG): Messung der elektrischen Muskelaktivität
Elektroneurographie (ENG): Messung der Nervenleitgeschwindigkeit
spinales und kraniales MRT: Scans von Gehirn und Wirbelsäule
Liquordiagnostik: Untersuchung des Hirnwassers
Blutuntersuchungen: BSG, CRP, Blutbild, GOT, GPT, TSH, Vitamin B12, Elektrophorese, CK, Kreatinin, Elektrolyte, Glukose
Laborwerte: Ausschluss von Autoimmunerkrankungen oder bakteriellen Infektionen wie Borreliose oder Syphilis (Lues)
genetische Diagnostik: bei Verdacht auf familiäre Veranlagung
Zur Differenzialdiagnostik und Einschätzung der Prognose ist eine Untersuchung von Liquor und Blutserum auf Neurofilamente (NFL) möglich. Diese Proteine, bilden das Gerüst von Nervenzellen. Gehen Neurone wie bei ALS zugrunde, wird NFL freigesetzt und kann als Biomarker verwendet werden.
Wie kann ALS behandelt werden?
Bisher gibt es keine Therapie, die ALS heilen kann. Daher konzentriert sich die Behandlung darauf, Symptome zu lindern und die Lebensqualität zu steigern. Empfehlenswert ist die Beratung in einer auf ALS spezialisierten Klinik.
Mit Riluzol steht ein Wirkstoff zur Verfügung, der das Fortschreiten der Nervenläsionen eindämmen kann. Das Medikament verlangsamt den Krankheitsprozess und verlängert die durchschnittliche Überlebenszeit um etwa drei bis vier Monate. Mögliche Nebenwirkungen von Riluzol sind Übelkeit, Erbrechen, Durchfall, Müdigkeit und erhöhte Leberwerte.
Zur symptomatischen Therapie der Beschwerden stehen verschiedene Wirkstoffe zur Verfügung:
- Antibiotika bei Lungenentzündung
- Schmerzmittel gegen Muskelschmerzen
- Magnesium und Chininsulfat gegen Muskelkrämpfe
- schleimlösende Medikamente wie N-Acetylcystein
- zur Hemmung der Speichelbildung Anticholinergika, eventuell auch Injektionen mit Botolinumtoxin (Botox) in die Speicheldrüsen
Allgemeine Maßnahmen zur Therapie
Erfolgversprechend ist im Anfangsstadium Physiotherapie zum Erhalt der Beweglichkeit. Ergänzend können Ergotherapie und bei Sprach- und Schluckproblemen Logopädie sehr hilfreich sein. Bei eingeschränkter Beweglichkeit kommen Hilfsmittel wie Gehstock, Rollator und Rollstuhl zum Einsatz, um möglichst lange die Selbständigkeit der Patient*innen zu erhalten.
Therapie im fortgeschrittenen Stadium
Im fortgeschrittenen Krankheitszustand stehen lebenserhaltende Maßnahmen wie Beatmung und künstliche Ernährung im Vordergrund:
Atemprobleme behandeln: Zunächst können Atemgymnastik, Klopfmassagen und schleimlösende Maßnahmen ergriffen werden. Später ist womöglich eine Beatmung mit einem mobilen Beatmungsgerät und einer Atemmaske notwendig. Ist die Atemmuskulatur so schwach, dass die Lunge nicht mehr ausreichend belüftet wird, muss auf maschinelle Beatmung umgestellt werden.
Magensonde: Die Schluckstörungen führen häufig zu Mangelernährung und Dehydration. Durch eine Magensonde kann hochkalorische, flüssige Nahrung zugeführt werden.
Sprachcomputer: Sie erleichtern die Kommunikation bei Sprechstörungen. Alternativ kann eine Bildertafel verwendet werden, auf der Buchstaben, Zahlen oder wichtige Begriffe aus der Pflege (etwa Schmerzintensität oder Bedürfnisse wie Hunger und Durst) dargestellt sind.
Psychotherapie: Aufgrund der Schwere der Krankheit kommt es gerade im Anfangsstadium zu Ängsten und Depressionen. Es ist wichtig, dass Betroffene und Angehörige professionelle psychotherapeutische Unterstützung erhalten. Teilweise können psychische Symptome auch durch Medikamente wie Antidepressiva gebessert werden.
Verlauf, Prognose und Lebenserwartung bei ALS
Bei einer klassischen ALS ist die Lebenserwartung nicht besonders hoch. Nach wie vor gilt die ALS als unheilbar und die meisten Erkrankten versterben innerhalb von zwei bis fünf Jahren. Nur etwa 10 Prozent leben länger als fünf Jahre, etwa fünf Prozent länger als zehn Jahre. Besser ist die Lebenserwartung allerdings bei den verschiedenen Sonderformen.
Trotz der schweren Erkrankung und der insgesamt schlechten Prognose ist auch in fortgeschrittenem Krankheitsverlauf häufig eine relativ gute Lebensqualität möglich.
Todesursache bei ALS
Schlussendlich führt bei den meisten Patient*innen das Aussetzen der Atemmuskulatur zum Tod. Fachleute sprechen von zunehmender respiratorischer Insuffizienz. Die Schwächung der Atemmuskeln führt in erster Linie nicht zum Sauerstoffmangel, sondern zu einem Überschuss an Kohlendioxid, was die Betroffenen zunehmend müde macht. Insofern kommt es bei einer ALS normalerweise eher zum langsamen Einschlafen.

Bewegungsfähigkeit und Ruhezittern sind die typischen Symptome von Parkinson. Was lässt sich gegen die Schüttellähmung tun? → Weiterlesen
Bei Muskelschwund schrumpfen die Muskeln eines Menschen. Was kann dahinter stecken und was hilft? → Weiterlesen
Die Gehirnerkrankung wird durch einen Gendefekt ausgelöst und ist unheilbar → Weiterlesen
Viele ADHS-Kinder leiden auch als Erwachsene noch unter der Störung. Woran man sie erkennt → Weiterlesen
Morbus Alzheimer ist die häufigste Demenzform und bis heute unheilbar. Die Symptome der Krankheit lassen sich in drei Stadien einteilen → Weiterlesen
Typisch für Epilepsie sind Zuckungen und Krampfanfälle. Alles über Ursachen, Symptome, Erste Hilfe und Therapie! → Weiterlesen
Was ist eine Aphasie, was sind die Ursachen und wie wird behandelt? Außerdem Tipps für Angehörige → Weiterlesen
Muskeldystrophie Duchenne führt zu fortschreitender Muskelschwäche und verkürzt die Lebenserwartung. → Weiterlesen
Sie möchten Informationen zu bestimmten Krankheitssymptomen oder wollen medizinischen Rat? Hier können Sie Ihre Fragen an unsere Experten oder andere Lifeline-Nutzer stellen!



